

生命科学は、情報学・数学・1分子計測技術の魅力的な実験場です。私たちは、これらの学問を生命科学に取り入れ、DNAとオーミクス周辺の生命科学の研究課題に取り組んでいます。
情報学・数学からのアプローチ
数学的思考等の知的活動を計算機で自動化することには限界があることは、たとえばゲーデルの不完全性定理等で知られています。この限界を意識しつつ、自動化できる範囲が機械学習、データマ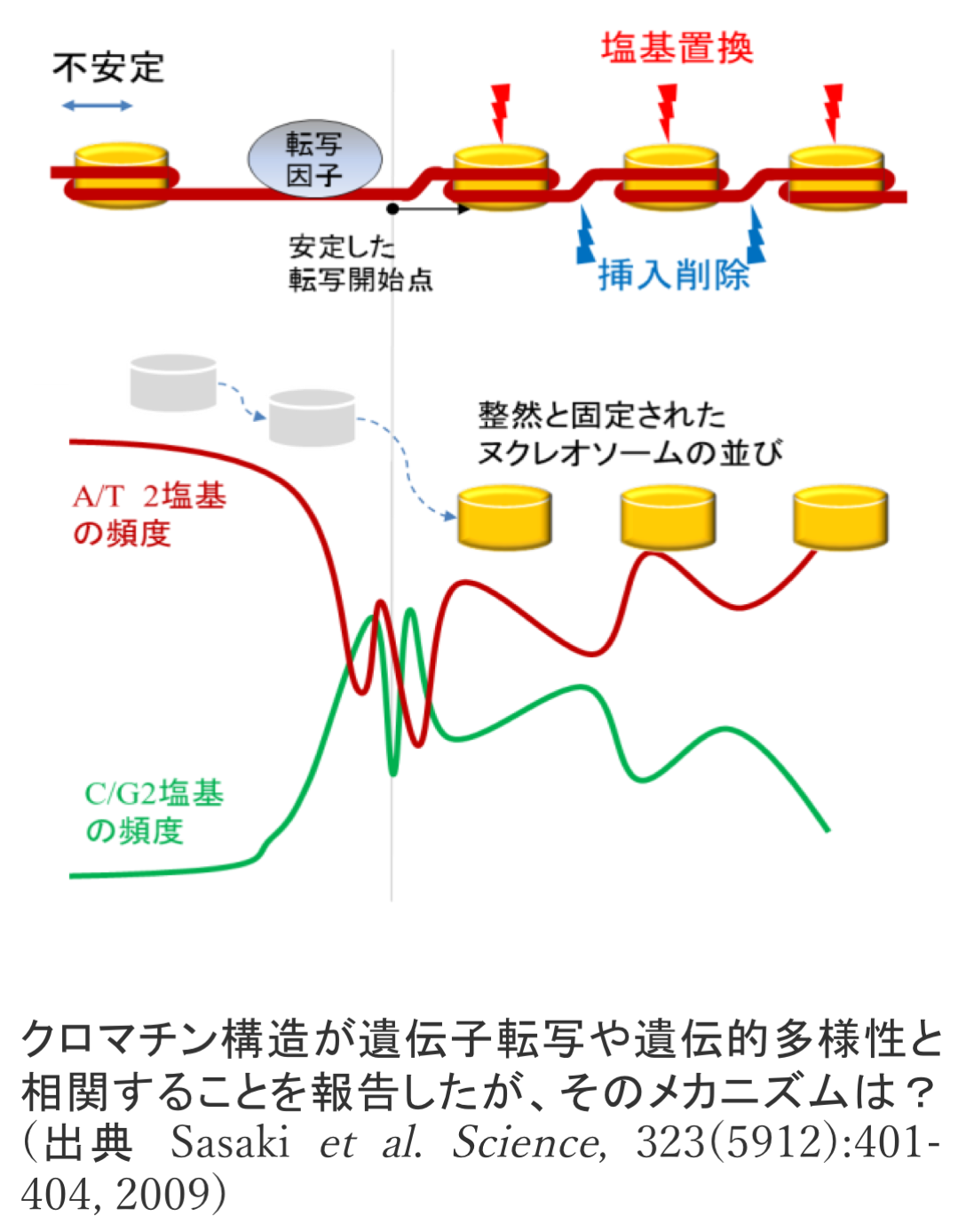 イニング等の情報学分野で徐々に広がっています。一方、私たちが取り組んでいるのは、生物における自動化メカニズムの理解です。たとえば、ゲノムがどのように情報をコードして細胞や組織を構築し、また進化してきたか? 核内でDNAはどのように畳み込まれ遺伝子の発現を制御しているか? などを深く知りたいと考えています。このような歩みから、生命科学は情報学と数学の魅力的な実験場であると感じています。特に最適化アルゴリズム、並列計算、機械学習、定理証明技術、計算幾何学 代数幾何学は、生命における自動化の仕組みを分析することに役立ってきています。
イニング等の情報学分野で徐々に広がっています。一方、私たちが取り組んでいるのは、生物における自動化メカニズムの理解です。たとえば、ゲノムがどのように情報をコードして細胞や組織を構築し、また進化してきたか? 核内でDNAはどのように畳み込まれ遺伝子の発現を制御しているか? などを深く知りたいと考えています。このような歩みから、生命科学は情報学と数学の魅力的な実験場であると感じています。特に最適化アルゴリズム、並列計算、機械学習、定理証明技術、計算幾何学 代数幾何学は、生命における自動化の仕組みを分析することに役立ってきています。
- 線虫ゲノムの trans-splicing 構造の解明 [Genome Res. 2013]
- DNAメチル化が影響する新たな遺伝的多様性 [Genome Res. 2012]
- クロマチン構造が影響する新たな遺伝的多様性 [Science 2009]
定理の証明を理解し、アルゴリズムを実装することが大事です。生命科学で新しい結果が出た場合は科学系の雑誌に報告し、一方、コンピュータ・サイエンスとして価値がある場合には ACM, IEEE 等の雑誌に報告してきました。
- Computer Science での主な論文リスト(後半に書いてあります)
1分子・1細胞を計測する技術の開拓
新しい観測技術が新しい発見を導くことが多いことはご存知かと思います。1分子や1細胞の動きを直接観測する技術はそのような例です。1分子計測技術を研究開発しているシリコンバレーの企業Pacific Biosciences社と、私たちは長年共同で研究して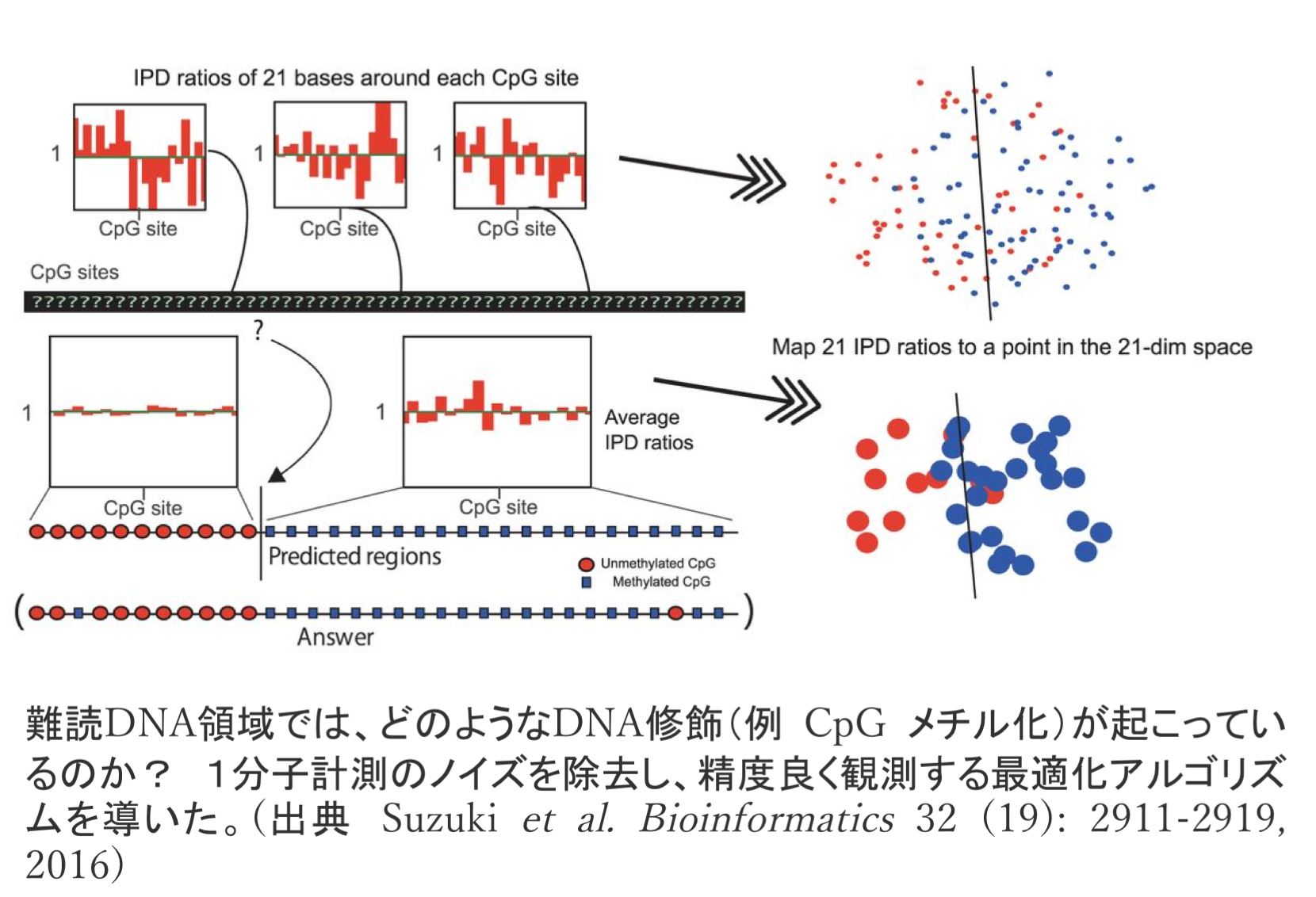 います。同社のZero-Mode Waveguide (ZMW) 技術は1分子計測の傑作で、たとえばDNAポリメラーゼの動きを実時間で直接観測できるため、これまで観測が困難だった難読領域のCpG メチル化を測定するアルゴリズムを一緒に研究開発しました。
います。同社のZero-Mode Waveguide (ZMW) 技術は1分子計測の傑作で、たとえばDNAポリメラーゼの動きを実時間で直接観測できるため、これまで観測が困難だった難読領域のCpG メチル化を測定するアルゴリズムを一緒に研究開発しました。
現在は、メタゲノム(細菌叢)および多細胞生物(線虫、ヒト等)で、ZMWを用いて多様なDNA修飾(6mA, 4mC等)を観測し、それらの機能を分析しています。
- ZMWの1分子計測ノイズを最適化アルゴリズムおよび機械学習により除去し、CpG メチル化難読領域を観測する技術を研究開発しました。これまで観測が困難なゲノムセントロメア領域や繰返し配列(Alu, LINE, LTR)のCpG メチル化、相同染色体間のアレル特異的メチル化の測定が可能になりました。[Bioinfo. 2016]
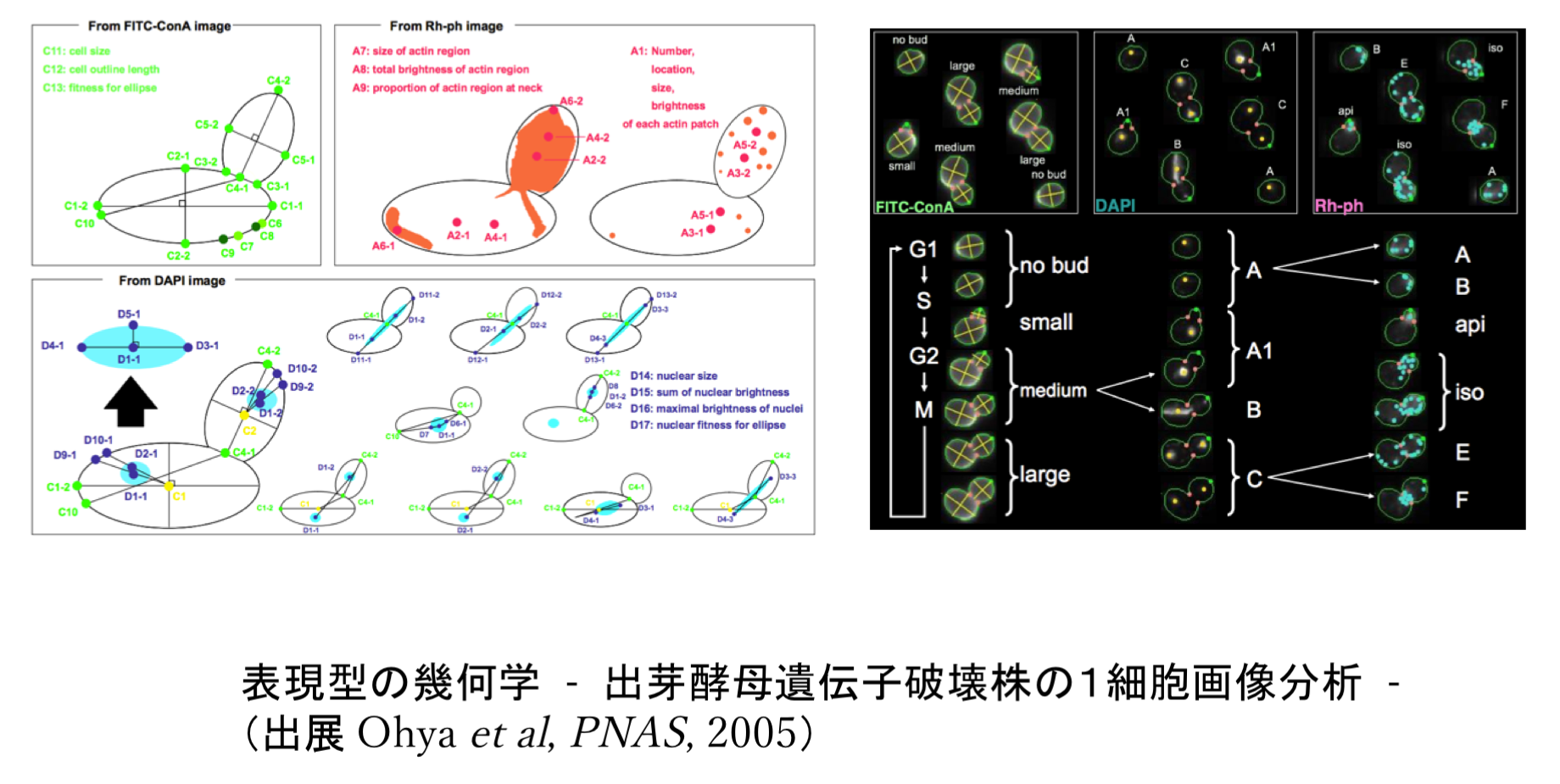
- 出芽酵母遺伝子破壊株の1細胞画像解析 [PNAS 2005]
DNAとオーミクス周辺の生命科学の研究課題
21世紀に入り、ヒトを始めとする巨大DNAが解読されるようになり、いろいろな謎が解明されています。たとえば遺伝学者の大野は、脊椎動物祖先で2回の全ゲノム重複が起こったと1970年に予測しました(大野の仮説)。この全ゲノム重複が遺伝子集合を豊かにし、脊椎動物の繁栄をもたらしたと考えられています。私たちは魚類(メダカ)ゲノムを解読し、哺乳類ゲノムを比較することにより、大野の仮説を立証しました [Nature, Genome Res 2007]。
- 脊椎動物の全ゲノム重複(大野の仮説)の立証 [Genome Res 2007]
その後は、ヒト個人ゲノムの解読に力点を移し、東大病院にて約5千人の個人ゲノムを解読し、脳疾患および癌に関連する遺伝子変異を分析しています。このような解析は、一昔前までは難しかったのですが、現在では容易になりました。いま取り組んでいるのは、解読困難なDNA領域の解明です。難読なセントロメア配列、ヒト腸内細菌叢、個人特異的な繰返し配列(LINE, LTR等)などを研究対象にしています。最先端のDNAシーケンサー(第三世代・長鎖型PacBio Sequel, Oxford nanopore, 10X Chromiumおよび次世代・短鎖型Illumina HiSeq)を利用し、研究室内でDNAデータを収集してます。特に、数万塩基の長いDNA断片を解読できる長鎖型は、謎の理解に役立っています。
- セントロメア配列の解明と、セントロメアDNAメチル化
- ヒトおよび霊長類ゲノム上の繰返し配列(LINE, LTR等)のDNAメチル化解析
- ヒト腸内細菌叢の全ゲノム、全メチル化、全プラスミド・ファージの解析
- 東大病院にて約5千人の個人ゲノムを解読し、脳疾患および癌に関連する遺伝子変異を多数報告
- 大規模構造変異を収集し、それらが生む新しい遺伝子転写制御を解析
大学院生の研究テーマ選び
自分が興味をもった研究でないと長続きしないので、研究課題の選択は各自に委ねています。ただ新しい課題でないと困るし、インパクトもほしいので、研究室全体で議論します。研究室に無いデータ、サンプル、装置が必要なとき共同研究します。ほとんどの大学院生が、学内の研究室、理研、スタンフォード大学、ワシントン大学等の海外の大学と共同研究してます。英語でメールすることから始めて、国際会議に参加し、共同研究先に訪問・滞在し、実践的な英語力を高めています。